
Introduction
Ischemic stroke is caused primarily by an interruption in cerebral blood flow, which induces severe neural injuries, and is one of the leading causes of death and disability worldwide. Thus, it is of great necessity to further detailly elucidate the mechanisms of ischemic stroke and find out new therapies against the disease. In recent years, efforts have been made to understand the pathophysiology of ischemic stroke, including cellular excitotoxicity, oxidative stress, cell death processes, and neuroinflammation. In the meantime, a plethora of signaling pathways, either detrimental or neuroprotective, are also highly involved in the forementioned pathophysiology. These pathways are closely intertwined and form a complex signaling network. Also, these signaling pathways reveal therapeutic potential, as targeting these signaling pathways could possibly serve as therapeutic approaches against ischemic stroke. In this review, the signaling pathways involved in ischemic stroke was described and categorize them based on the pathophysiological processes they participate in. Therapeutic approaches targeting these signaling pathways, which are associated with the pathophysiology mentioned above, are also discussed. Meanwhile, clinical trials regarding ischemic stroke, which potentially target the pathophysiology and the signaling pathways involved, are summarized in details. Conclusively, this review elucidated potential molecular mechanisms and related signaling pathways underlying ischemic stroke, and summarize the therapeutic approaches targeted various pathophysiology, with particular reference to clinical trials and future prospects for treating ischemic stroke.
Pathophysiology and signaling pathways involved in ischemic stroke
Energy deficiency due to a lack of glucose and oxygen
Immediately after ischemic stroke, cerebral blood flow is significantly reduced, which limits the availability of glucose and oxygen, especially in neurons. Energy disruption leads to mitochondrial dysfunction and oxidative stress-induced damage, triggered by the production of ROS. Concurrently, energy deficiency contributes to an ionic imbalance that affects Na+, K+, and Ca2+ levels, leading to cell depolarization and prompting glutamate release. The excessive glutamate activates N-methyl-D-aspartate receptors (NMDARs), inducing toxicity, cell death, and finally severe damage of the central nervous system. Taken together, deficiency in glucose and oxygen may eventually lead to cellular excitotoxicity and mitochondrial dysfunctions, which serve as the initiating session of ischemia-induced damage and subsequently cause other cascade of injuries. In this section, the review focuses on the various signaling pathways involved in glutamate and NMDAR-induced cell toxicity, namely, excitotoxicity, as well as oxidative stress and mitochondrial dysfunction in ischemic stroke.
Excitotoxicity and related signaling pathways
Glucose and oxygen deficiency during cerebral ischemia induces neuronal cell depolarization and glutamate release. The latter then stimulates Na+/Ca2+ channels coupled with NMDARs. Enhanced Ca2+ influx perturbs ionic homeostasis, resulting in Ca2+ overload in both the mitochondria and cytosol. These changes stimulate a variety of proteases, lipases, kinases, phosphatases, endonucleases, and free radicals, as well as biological processes causing cell death, such as calpain activation, oxidative stress, and mitochondrial impairment. Overall, these cellular dysfunctions are termed excitotoxicity and involve NMDARs, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, and kainite receptors.
Despite their involvement in ischemic stroke-related excitotoxicity, NMDARs act as a double-edged sword. Functional and structural studies have revealed that activation of NMDARs containing the GluN2B subunit triggers excitotoxicity during ischemic stroke and subsequent neuronal apoptosis, whereas activation of NMDARs containing the GluN2A subunit exerts a neuroprotective effect. Similarly, it has been hypothesized that synaptic NMDARs promote neuronal survival, whereas extra-synaptic NMDARs play detrimental roles in neuronal activity. The analogy between synaptic vs. extra-synaptic NMDARs and GluN2A-containing NMDARs vs. GluN2B-containing NMDARs demonstrates the dual effect of NMDARs and their regulation of signaling pathways with neuroprotective or detrimental effects on ischemic stroke (Fig. 1)
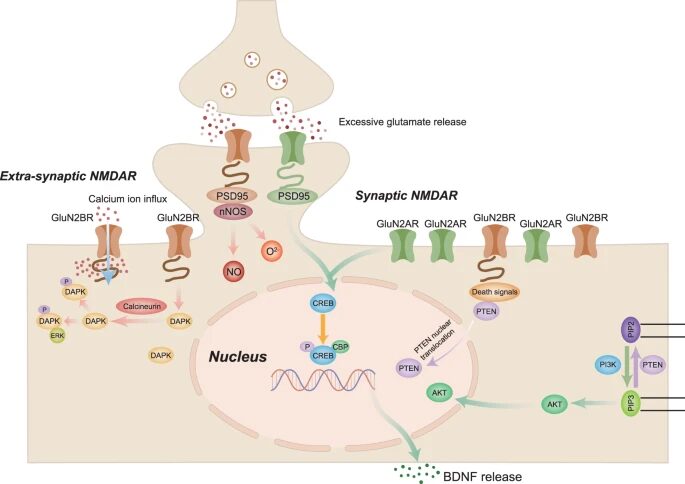
Figure 1. Excitotoxicity and signaling pathways involved in ischemic stroke. NMDAR N-methyl-D-aspartate receptors, PI3K Phosphatidylinositol 3 kinase, BDNF Brain-derived neurotrophic factor, CREB cAMP-response element-binding protein PTEN Phosphate and tension homology deleted on chromosome ten, PIP3 plasma membrane intrinsic protein 3, DAPK1 Death-associated protein kinase 1, PSD95 Postsynaptic density protein 95, nNOS Neuronal nitric oxide synthase. Adapted from source
Phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway
Stimulation of synaptic NMDARs activates the pro-survival PI3K/Akt signaling pathway, thereby exerting a neuroprotective effect. PI3K is an intracellular kinase classified into three categories (I, II, and III) based on structure and substrate specificity. In neurons, activation of the PI3K/Akt signaling pathway by NMDAR occurs via Ca2+ and calmodulin, which recruit phosphoinositide-dependent protein kinase 1. At the same time, Ca2+ triggers tyrosine phosphorylation of insulin receptor substrate 1, reinforcing NMDAR-induced Akt activation. The protective effect of the PI3K/Akt signaling pathway on ischemic stroke has been reported both in in vitro neurons during hypoxia and in vivo against ischemic neuronal death, and PI3K/Akt signaling inhibition aggravates ischemia-induced neuronal death in experimental stroke animals. Mechanistically, the neuroprotective effect of Akt is related to the phosphorylation and inactivation of various downstream targets, including glycogen synthase kinase 3 beta (GSK3β), pro-apoptotic B-cell lymphoma 2 (Bcl2)-associated BAD, c-Jun N-terminal kinase (JNK)/p38 activator ASK1, and apoptotic p53. These effects not only exist in neurons, but also in other neural cell types, which are possibly related to the inhibition of synaptic excitotoxicity and thus exert neuroprotective effects in ischemic stroke.
Brain-derived neurotrophic factor (BDNF) and cAMP-response element-binding protein (CREB)-related gene products
Synaptic NMDAR activation and Ca2+ influx activate the Ras/extracellular signal-regulated kinase (ERK) signaling pathway and nuclear Ca2+/calmodulin-dependent protein kinases, which in turn phosphorylate and activate CREB. Together with NMDAR and BDNF, CREB promotes the expression of numerous pro-neuronal survival genes. BDNF production in the brain relies on Ca2+ influx through NMDAR. Synaptic NMDARs promote BDNF gene expression, whereas extra-synaptic NMDARs block CREB-mediated BDNF expression. In experimental ischemic stroke models, BDNF is secreted into the brain and protects against ischemia-induced injury via neuronal GluN2A-NMDAR activation. Together, these results show that BDNF and, to some extent, the upstream CREB signaling pathway contribute to the neuroprotective effect associated with synaptic excitotoxicity in cerebral ischemia.
Phosphatase and tensin homolog (PTEN) signaling pathway
Extra-synaptic NMDARs are closely linked to signaling pathways associated with cell death and often contradict the effects triggered by synaptic NMDARs. Upon activation by Ca2+ influx through NMDARs, PTEN is recruited to GluN2B-NMDARs. The direct interaction between PTEN and the GluN1 subunit of GluN2B-NMDARs enhances current flow through the channel, tightening the junctions between PTEN and the neuronal death signaling complex. Concurrently, the excitotoxic stimulation of NMDARs initiates PTEN nuclear translocation, thus significantly lowering the phosphorylation of phosphatidylinositol-trisphosphate and Akt and consequently blocking PI3K/Akt signaling. Thus, contrary to the protective effect of PI3K/Akt, PTEN signaling may decrease cell survival and induce neuronal death. In agreement with this hypothesis, downregulating PTEN expression reportedly inhibits extra-synaptic NMDAR currents and protects neurons from experimental ischemic injury. The above evidence hints at the detrimental role of PTEN in ischemic stroke, which is largely mediated by regulation of extra-synaptic NMDAR activities.
Death-associated protein kinase 1 (DAPK1) signaling pathway
DAPK1 is a Ca2+/calmodulin-dependent serine/threonine-protein kinase, whose phosphorylation contributes to apoptotic cell death. DAPK1 participates in excitotoxicity in ischemic stroke. During ischemia, NMDAR overactivation promotes Ca2+ influx, activates Ca2+/calmodulin, and stimulates calcineurin phosphatase, which subsequently dephosphorylates and activates DAPK1. The latter is then transferred to the GluN2B subunit of NMDARs, aggravating ischemic injury. Preventing the interaction between GluN2B and DAPK1 attenuated neuronal excitotoxicity in mouse ischemic stroke models and downregulated the NMDAR current in vitro. In addition, NMDAR-regulated calcineurin activation contributes to DAPK1 activation, whereas NMDAR or calcineurin inhibition prevents DAPK1 dephosphorylation. DAPK1 inhibition protects against ischemic injury both in cultured neurons and in vivo, suggesting that potential treatments for ischemic stroke could be based on inhibiting DAPK1. It is interesting to note that the pro-survival signaling factor ERK serves as a downstream effector of DAPK1, and the DAPK1-ERK interaction could block the neuroprotective effect of ERK on experimental ischemic stroke, possibly by retaining ERK in neuronal cytoplasm.
Postsynaptic density protein-95 (PSD95)/neuronal nitric oxide synthase (nNOS) signaling pathways and excitotoxicity-induced cell death
Neuronal NMDARs contribute to nitric oxide production, which is associated with calcium/calmodulin and is regulated by nNOS. NMDAR subunits bind directly to PSD95, which is composed of three PDZ domains. The binding of PSD95 to NMDAR and nNOS enhances Ca2+ influx, a hallmark of excitotoxicity. PSD95/nNOS signaling may play a pivotal role in ischemic stroke, as evidenced by the amelioration of neurological deficits in animals suffering from cerebral ischemia and whose nNOS activity was inhibited by either pharmacological or genetic means. Cerebral ischemia has been shown to enhance NMDAR/PSD95/nNOS interactions in neurons, thus further aggravating brain injuries after experimental ischemic stroke. All these results show that signaling through the PSD95/nNOS complex is crucial for excitotoxicity in ischemic stroke and contributes to the neurotoxic effects of extra-synaptic NMDARs.
Mitochondrial dysfunction, oxidative stress, and related signaling pathways
Mitochondria are essential for maintaining energy homeostasis. When ATP synthesis and energy balance are disrupted by a lack of glucose and oxygen, the status and function of mitochondria become substantially altered. Ca2+ influx leads to mitochondrial permeability transition pore (MPTP) opening and cytochrome c release. At the same time, insufficient ATP supply triggers mitochondrial membrane depolarization, which is characterized by the influx of Na+ and efflux of K+. Besides mitochondrial dysfunction, energy deficiency in cerebral ischemia leads to oxidative stress, which severely damages cells and brain tissues. Oxidative stress accompanies several pathological processes and results from increased ROS production, mostly via oxidative phosphorylation in the mitochondria. Considering the intimate link between ROS and mitochondrial metabolism, mitochondrial dysfunction is often related to oxidative stress pathologies. During ischemia, oxidative damage and excessive Ca2+ levels contribute to MPTP induction, which further promotes succinate release and mitochondrial damage-associated molecular patterns including the activation of downstream inflammatory responses. Consequently, all these damaging factors lead to neurotoxic and cell death processes, in which a plethora of signaling pathways are involved (Fig. 2).
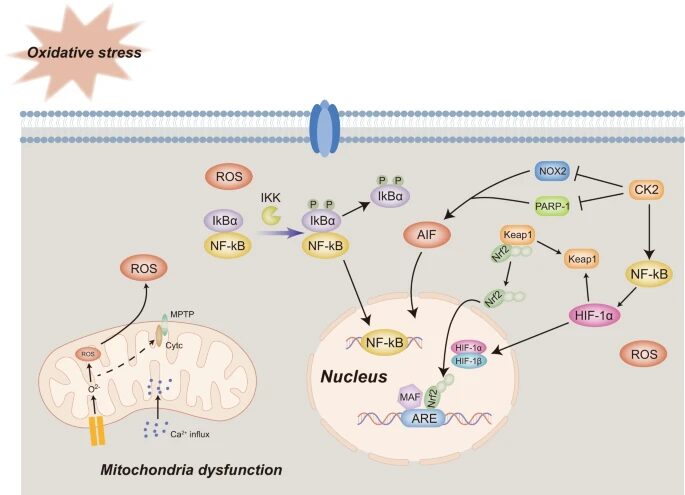
Figure 2. Oxidative stress and mitochondrial dysfunctions and signaling pathways involved in ischemic stroke. MPTP mitochondrial permeability transition pore, ROS Reactive oxygen species, ATP Adenosine triphosphate, HIF-1 Hypoxia-induced factor, Nrf2 Nuclear factor E2-related factor 2, ARE Antioxidant response element, CK2 Casein kinase 2, PARP-1 Poly ADP-ribose polymerase 1, AIF Apoptosis-inducing factor, PINK1 PTEN induced putative kinase 1, NF-kB Necrosis factor-kB. . Adapted from source
Concluding remarks and future perspectives
Ischemic stroke is characterized by the blockade of cerebral blood flow caused by the presence of thrombi in the blood vessels and has an overwhelming effect on people’s health and their quality of life. In recent years, studies have sought to further elucidate the mechanisms of ischemic stroke. Nevertheless, the complex pathogenesis of ischemic stroke means that the participating signaling pathways need further comprehensive exploration. In this review, the signaling pathways involved in ischemic stroke was summarized and categorized them based on their specific pathophysiological roles in excitotoxicity, mitochondrial dysfunction, oxidative stress, neuroinflammation, and cell death. Because these signaling pathways are interconnected, combined therapeutic targets against ischemic stroke may be elucidated.
At present, recanalization of blood vessels via intravenous thrombolytic treatment or mechanical thrombectomy represents the major therapeutic approach for ischemic stroke. However, this is underscored by the lack of suitable pharmacological treatments, calling for the discovery of new therapeutic targets against ischemic stroke. In this review, through existing therapeutic approaches was combined and classified them according to their target signaling pathways.