

Introduction
Inhibition of chaperone-mediated autophagy (CMA), a selective type of lysosomal degradation for intracellular proteins, may contribute to pathogenesis in neurodegenerative diseases including Parkinson disease (PD). Pathogenic variants of PD-related proteins that reside in the cytosol, including SNCA/alpha-synuclein, LRRK2 (leucine rich repeat kinase 2), UCHL1 (ubiquitin C-terminal hydrolase 1) and VPS35 (VPS35 retromer complex component), exert inhibitory effects on CMA. Decreased CMA activity has also been reported in sporadic PD patients, consistent with an association between CMA inhibition and PD. The first example of CMA dysfunction was reported caused by a non-cytosolic PD-related protein, GBA/β-glucocerebrosidase, the most common genetic risk factor for PD, which uncovers a new role for CMA in endoplasmic reticulum (ER) quality control.
GBA gene
GBA is a lysosomal enzyme that reaches the lysosomal lumen after synthesis in the ER and trafficking in vesicles secreted from the Golgi (Figure 1(a)). Patients with the lysosomal storage disorder Gaucher disease possess two mutant alleles of the GBA/GBA1 gene, and because they lack any enzymatic activity, in many cases, enzyme replacement provides successful treatment. In contrast, in PD, heterozygous mutant alleles of the GBA gene encode mutant (mt) forms of GBA that fail to properly fold in the ER. In the recent study, using an array of disease-relevant models including primary ventral midbrain dopamine neurons from transgenic mouse models expressing mtGBA (GBAL444P), fibroblasts, and induced pluripotent stem cells (iPSCs) derived into dopaminergic neurons from patients with heterozygous GBAWT/N370S mutations (GBA-PD), accumulation of mtGBA was observed in the ER and low levels of the mutant enzymes within lysosomes. Unexpectedly, isolation of lysosomes from postmortem GBA-PD patient brains revealed a topological misplacement of the enzyme with a fraction of mtGBA detected on the lysosomal surface instead of the lumen. This mistrafficked GBA was confirmed in the various GBA-PD experimental models where it also demonstrated reduced CMA activity. Using pharmacological inhibitors of GBA, it was found that CMA inhibition is independent of GBA enzymatic activity. Importantly, however, the CMA blockade is prevented by eliminating a CMA targeting motif (256QRDFI260) in GBA.
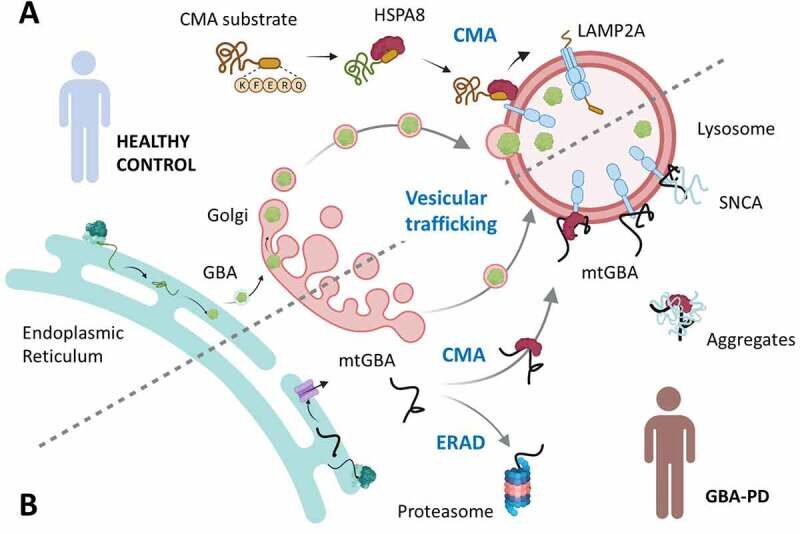
Figure 1. Disruptive effect of mutant GBA on chaperone-mediated autophagy (CMA) in Parkinson disease. (a) Normal delivery of folded functional GBA into lysosomes through ER-Golgi vesicular trafficking. Functional CMA is depicted as the targeting by HSPA8 of CMA substrate proteins to the CMA lysosomal receptor LAMP2A and subsequent internalization of the unfolded substrate through the CMA translocation complex. (b) Failure to fold mutant GBA (mtGBA) in GBA-PD, leads to its retrotranslocation into the cytosol where it can undergo degradation through the proteasome by ER-associated degradation (ERAD) or be targeted to lysosomes via CMA. Persistent binding of mtGBA to LAMP2A and its inhibitory effect on the formation of the LAMP2A translocation complex leads to CMA blockage and accumulation of other CMA substrates such as SNCA. Adapted from source
CMA
Degradation of cytosolic proteins by CMA involves the recognition of a pentapeptide targeting motif by the chaperone HSPA8/hsc70 that targets the substrate protein to the lysosomal membrane for binding to the CMA receptor LAMP2A (lysosomal associated membrane protein 2A). Once bound, the substrate protein is unfolded and internalized through a translocation complex and degraded within lysosomes. Wild-type (WT) GBA is not normally present in the cytosol, but mtGBA was detected in the cytosolic fraction of GBA-PD experimental models and patient brain. ER retrotranslocation of unfolded mtGBA proteins and subsequent degradation by the proteasome have been previously reported. In this work, it demonstrate recognition by HSPA8 of the 256QRDFI260 sequence in unfolded mtGBA that reaches the cytosol after ER retrotranslocation and its targeting to lysosomes for degradation by CMA, indicating an alternate degradation pathway for misfolded proteins in the ER.
Misfolded GBA is a poor CMA substrate, and despite strong binding to LAMP2A, exhibits slower translocation into the lysosomal lumen than other known substrates. In the case of the small fraction of WT GBA that fails to fold, the proteasome and CMA may successfully handle it. However, the higher levels of unfolded mtGBA that reaches the cytosol together with the inefficient lysosomal internalization through the CMA translocation complex, prevents GBA from degradation and also blocks degradation of other cytosolic proteins that rely on CMA for clearance (Figure 1(b)). These proteins include SNCA, the primary component of the protein inclusions known as Lewy bodies, the major PD pathological hallmark. Strikingly, results from the study in primary dopaminergic neurons indicate that the coincidence of mtGBA and SNCA produces neurotoxicity, and that either preventing CMA targeting of mtGBA or eliminating SNCA is sufficient for neuronal survival.
Support for a role for CMA dysfunction as a consequence of mtGBA in PD comes from proteomic analysis of postmortem brains and iPSC-derived dopaminergic neurons from GBA-PD patients. It was found that both types of GBA-PD patient samples display proteome changes in pathways related with cellular response to proteotoxicity, consistent with ER stress and protein misfolding problems. To characterize proteostasis abnormalities induced by GBA mutations, proteins that accumulate in the GBA-PD brain were analyzed and found marked enrichment of proteins bearing CMA motifs, indicating that blockade of CMA contributes to their intracellular accumulation. Last, comparative proteomics of the GBA-PD patients’ brains with brains of mice deficient in CMA in neurons (generated by knocking out Lamp2a) revealed a striking overlap of changes in the proteome of both samples.
Conclusion
Data from thi study supports a hypothesis that GBA mutations lead to misfolding and mislocalization of GBA to the cytosol, resulting in a toxic gain of function due to blockade of CMA. The subsequent accumulation of CMA substrates can be additional important contributors to GBA-PD pathogenesis. This finding differs from hypotheses that PD pathology due to GBA mutations results from insufficient enzymatic activity as in Gaucher disease. Instead, these studies support the idea that replacing the missing enzyme may not be sufficient to completely normalize cellular dysfunction in GBA-PD, at least for those mtGBA forms that inhibit CMA.
For GBA-PD, interventions aiming at enhancing proper folding of mutant GBA and/or improving CMA function could be appealing strategies. Because CMA dysfunction has been identified in various forms of genetic PD and idiopathic PD, CMA failure could provide a convergent pathway for many types of PD and explain why seemly unrelated protein mutations lead to a final common SNCA pathology and selective vulnerability of dopaminergic neurons in PD. Enhancing CMA could have a broader impact in treating PD and protein levels of CMA substrates may serve as biomarkers to monitor target engagement, although further validation is needed for future translational research.